ПОБ/ПООБ
Утвержден и введен в действие
Приказом Федерального
агентства по техническому
регулированию и метрологии
от 20 сентября 2017 г. N 1182-ст
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ
ФАРМАКОНАДЗОР. ПЕРИОДИЧЕСКИЕ ОТЧЕТЫ О БЕЗОПАСНОСТИ
ЗАРЕГИСТРИРОВАННЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Medicines for medical applications. Pharmacovigilance.
Periodic reports on the safety of registered drugs
(ICH E2C Periodic benefit-risk evaluation report
(PBRER), IDT)
ГОСТ Р 57690-2017
ОКС 11.120
Дата введения
1 августа 2018 года
Предисловие
- ПОДГОТОВЛЕН Государственным бюджетным образовательным учреждением высшего профессионального образования — Первым Московским государственным медицинским университетом имени И.М. Сеченова Министерства здравоохранения Российской Федерации (Первым МГМУ имени И.М. Сеченова)
- ВНЕСЕН Техническим комитетом по стандартизации 458 «Разработка, производство и контроль качества лекарственных средств»
- УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 20 сентября 2017 г. N 1182-ст
- Настоящий стандарт идентичен международному документу ICH E2C по подготовке периодических отчетов об оценке риска-пользы лекарственных средств [ICH E2C Periodic benefit-risk evaluation report (PBRER)] Международной конференции по гармонизации технических требований для регистрации лекарственных средств для медицинского применения (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; ICH) <1>
———————————
<1> С 25 октября 2015 г. переименована в Международный совет по гармонизации.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Оглавление
1.3. Цели периодического отчета по безопасности. 5
1.4. Принципы оценки соотношения польза-риск в ПОБ. 6
2.2. Требования к содержанию каждой части ПОБ. 11
2.3 Система качества ПОБ на уровне держателя регистрационных удостоверений 39
2.4. Обучение персонала процедурам по ПОБ. 41
2.5. Порядок представления ПОБ. 42
Введение
Данный стандарт направлен на определение общих требований по подготовке периодических отчетов о безопасности (о соотношении риск-польза, ПОБ) лекарственных препаратов, находящихся в обращении в странах ICH. Стандарт содержит рекомендуемый формат и содержание ПОБ, описание вопросов, требующих внимания, при подготовке и представлении в регуляторный орган данных отчетов.
Руководству ICH E2C по подготовке периодических отчетов об оценке риск-польза лекарственных средств (ICH E2C Periodic benefit-risk evaluation report (PBRER)) Международной конференции по гармонизации технических требований для регистрации лекарственных средств для медицинского применения (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; ICH).
1. Область применения
Область действия настоящего стандарта распространяется на все лекарственные препараты, находящиеся в обращении.
1.2 Общие принципы
Периодический отчет по безопасности (ПОБ) представляет собой документ по фармаконадзору, целью которого является представление держателем регистрационного удостоверения оценки соотношения польза/риск лекарственного препарата на определенных этапах пострегистрационного периода.
Регуляторными органами должна выполняться оценка ПОБ с определением возможных новых выявленных рисков и их влияния на оценку соотношения польза-риск лекарственного препарата. По результатам оценки регуляторный орган определяет необходимость выполнения дальнейших исследований/испытаний безопасности или эффективности лекарственного препарата, принятия регуляторных действий в отношении регистрационного статуса лекарственного препарата или внесения изменений в инструкцию по медицинскому применению лекарственного препарата в целях обеспечения его применения при превышении пользы над риском.
1.3. Цели периодического отчета по безопасности
Основной целью ПОБ является представление исчерпывающего и критического анализа соотношения польза-риск лекарственного препарата с учетом всех новых данных по безопасности и их кумулятивного влияния на профиль безопасности и эффективности лекарственного препарата. ПОБ является инструментом пострегистрационной оценки соотношения польза-риск лекарственного препарата на определенных этапах жизненного цикла лекарственного препарата.
В процессе пострегистрационного применения лекарственного препарата выявляется новая информация по безопасности, на основании оценки которой держателем регистрационного удостоверения должен постоянно выполняться анализ влияния новых данных на соотношение польза-риск, переоценка данного показателя, а также определяться необходимость оптимизации соотношения польза-риск путем введения эффективных мер по управлению рисками и их минимизации.
1.4. Принципы оценки соотношения польза-риск в ПОБ
Оценка соотношения польза-риск должна носить непрерывный характер на протяжении всего жизненного цикла лекарственного препарата в целях обеспечения защиты здоровья населения и повышения безопасности пациентов путем реализации эффективных мер минимизации риска. Основой для анализа является информация по безопасности и эффективности, собираемая на протяжении соответствующих промежутков времени, составляющих отчетные периоды. Оценка включает следующие этапы:
- Критический анализ всей информации по безопасности, полученной за отчетный период с определением возможных выявленных новых сигналов, свидетельствующих о новых потенциальных или идентифицированных рисках, либо дополнении имеющихся знаний по ранее идентифицированным рискам.
- Критическое обобщение всей полученной за отчетный период информации по безопасности и эффективности лекарственного препарата (как в рамках клинических исследований/испытаний, так и при применении в медицинской практике) с оценкой влияния этих данных на соотношение польза-риск лекарственного препарата.
- Выполнение интегрального анализа соотношения польза-риск на основании всех кумулятивных данных, имеющихся за период от даты первой регистрации в какой-либо из стран, даты первой регистрации для проведения интервенционного клинического исследования в какой-либо из стран.
- Обобщение информации по мерам минимизации риска, которые могли выполняться или планируются.
- Определение плана оценки сигналов и рисков и/или предложений по дополнительной деятельности по фармаконадзору.
2. Принципы подготовки ПОБ
Держатель регистрационного удостоверения должен готовить один ПОБ для всех своих лекарственных препаратов, содержащих одно и то же действующее вещество или одну и ту же комбинацию действующих веществ по всем одобренным показаниям, способам введения, формам выпуска и режимам дозирования. В определенных случаях может потребоваться представление данных по отдельным показаниям, формам выпуска, способам введения или режимам дозирования в отдельном разделе ПОБ с соответствующим отражением аспектов профиля безопасности, без подготовки отдельного ПОБ. В исключительных случаях подготовка отдельного ПОБ может быть обоснована, например, в случае отличной формы выпуска с полностью отличными показаниями к медицинскому применению.
2.1. Содержание ПОБ
ПОБ должен включать кумулятивные данные, полученные начиная от даты регистрации, с направленностью на новую информацию, полученную за отчетный период. Кумулятивная информация рассматривается при выполнении общей оценки безопасности и интегрированной оценки соотношения польза-риск.
ПОБ должен включать обобщающую информацию по всем источникам получения значимых данных по эффективности и безопасности, которые должны учитываться при выполнении очередной оценки соотношения польза-риск и которые имеются в распоряжении держателя регистрационного удостоверения. Указанная информация включает:
а) обобщающая информация по результатам медицинского применения:
— данные спонтанного репортирования;
— данные медицинской литературы;
— данные, полученные в ходе активных методов мониторинга (например, анализ внутренних или внешних бах данных);
— сигналы по безопасности, находящиеся на рассмотрении у держателя регистрационного удостоверения;
— информация от партнеров по маркетингу или дистрибьюции;
б) обобщающая информация по клиническим исследованиям/испытаниям:
— продолжающиеся клинические исследования/испытания или иные исследования/испытания, которые выполняются держателем регистрационного удостоверения или его представителем, либо которые были завершены в отчетный период;
— терапевтическое применение исследуемого лекарственного препарата;
— обсервационные или эпидемиологические исследования;
— исследования по оценке использования лекарственного препарата;
— доклинические исследования (токсикологические и исследования in vitro);
— клинические исследования, выполняемые партнерами держателя регистрационного удостоверения по разработке или размещению рынке лекарственного препарата;
— клинические исследования, в которых была выявлена недостаточная терапевтическая эффективность, что может оказать влияние на оценку соотношения польза-риск лекарственного препарата;
в) обобщающая информация из других источников:
— данные из иных источников, имеющих отношение к оценке эффективности или безопасности лекарственных препаратов аналогической фармакотерапевтической группы;
— иные ПОБ или отчеты по безопасности разрабатываемых лекарственных препаратов (например, контрактных партнеров или инициаторов исследований);
— важная информация, полученная после завершения подготовки ПОБ.
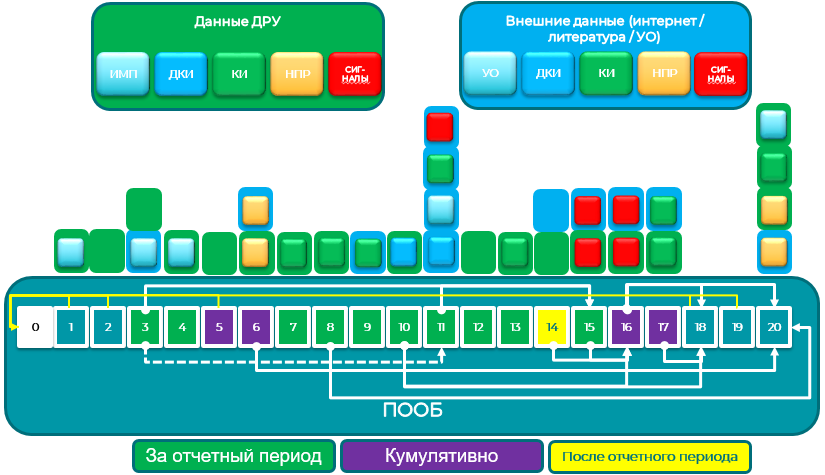
ПОБ должен включать следующие разделы:
Титульный лист, включая удостоверяющую подпись
Краткое изложение основного содержания
Таблица содержания отчета:
1) Введение
2) Регистрационный статус в мире
3) Меры, принятые за отчетный период, в связи с данными по безопасности
4) Изменения, внесенные в справочную информацию по безопасности лекарственного препарата
5) Оценка количества пациентов, подвергшихся воздействию лекарственного препарата
5.1 Общее количество пациентов, подвергшихся воздействию в клинических исследованиях
5.2 Общее количество пациентов, подвергшихся воздействию по данным применения на рынке
6) Обобщенные табличные данные
6.1 Справочная информация
6.2 Обобщенная информация по серьезным нежелательным реакциям, выявленным в ходе клинических исследований
6.3 Обобщенная информация по данным пострегистрационного применения
7) Резюме важных данных, полученных в ходе клинических исследований за отчетный период
7.1 Завершенные клинические исследования
7.2 Продолжающиеся клинические исследования
7.3 Длительный последующий мониторинг
7.4 Иное терапевтическое применение лекарственного препарата
7.5 Новые данные по безопасности в отношении назначения фиксированных комбинаций
8) Данные неинтервенционных исследований
9) Данные других клинических исследований и из других источников
10) Данные доклинических исследований
11) Литература
12) Другие периодические отчеты
13) Недостаточная терапевтическая эффективность в контролируемых клинических исследованиях
14) Важная информация, полученная после завершения подготовки ПОБ
15) Обзор сигналов: новые, рассматриваемые и завершенные
16) Сигналы и оценка риска
Обобщающая информация по проблемам по безопасности
Оценка сигнала
Оценка рисков и новой информации
Характеристика рисков
Эффективность мер минимизации риска (если применимо)
17) Оценка пользы
Важная базисная информация по эффективности в ходе клинических испытаний и применения в медицинской практике
Новая выявленная информация по эффективности в ходе клинических испытаний и применения в медицинской практике
Характеристика пользы
18) Интегрированный анализ соотношения польза-риск по одобренным показаниям
Контекст соотношения польза-риск – медицинская потребность и важные альтернативы
Оценка процедуры анализа соотношения польза-риск
19) Заключение и действия
20) Приложения к ПОБ
Титульный лист
На титульном листе должно быть указание номера отчета (отчеты должны иметь последовательную нумерацию), наименование лекарственного препарата, международную дату регистрации, отчетный период (либо указание на внеочередной порядок подачи по запросу регуляторного органа), дату составления отчета, данные держателя регистрационного удостоверения и указание о конфиденциальности информации, включенной в ПОБ. Титульный лист должен быть заверен подписью.
Краткое изложение основного содержания
Целью краткого изложения содержания является краткое представление в обобщенном виде содержания и наиболее важной информации, составляющей периодический отчет по безопасности. Данный раздел должен включать следующую информацию:
а) введение, указание номера отчета и отчетного периода;
б) наименование лекарственного препарата, фармакотерапевтичский класс, механизм действия, показания к применению, форма выпуска, доза, способ введения;
в) оценка кумулятивного воздействия в ходе клинических исследований;
г) оценка интервала пострегистрационного применения и кумулятивного воздействия за этот период;
д) число стран, на территории которых разрешено применение лекарственного препарата;
е) обобщенная информация по оценке соотношения польза-риск;
ж) принятые и предлагаемые действия, связанные с аспектами профиля безопасности, включая существенные изменения в брошюру исследователя на этапе клинических исследований и в инструкцию по медицинскому применению на пострегистрационном этапе, либо иные меры минимизации риска;
з) заключения.
Раздел краткого изложения содержания отчета должен сопровождаться таблицей содержания периодического отчета по безопасности.
2.2. Требования к содержанию каждой части ПОБ
2.2.1. Раздел ПОБ «Введение»
Введение должно содержать следующую информацию:
а) международная дата регистрации, отчетный период и порядковый номер отчета;
б) наименование лекарственного препарата, фармакотерапевтичский класс, механизм действия, показания к применению, форма выпуска, доза, способ введения;
в) краткое описание популяций, которые получают лечение с назначением лекарственного препарата или были включены в клинические исследования;
г) краткое описание и разъяснение любой имеющей отношение к требуемой информации в ПОБ, которая не была включена в подаваемый ПОБ.
2.2.2 Раздел ПОБ «Регистрационный статус в мире»
В данном разделе ПОБ должна быть представлена краткая обзорная информация, включающая даты первичных регистраций в странах мира, одобренные показания к применению, зарегистрированные формы выпуска и дозировки с указанием действующих на дату подготовки ПОБ регистраций.
2.2.3. Раздел ПОБ «Меры, принятые за отчетный период, в связи с данными по безопасности»
В разделе представляется описание существенных мер, принятых за отчетный период, как в отношении продолжающихся клинических исследований/испытаний, так и пострегистрационного применения, со стороны регуляторных органов, держателя регистрационного удостоверения, спонсора/заявителя клинических исследований, комитета по мониторингу/оценке данных, комитета по этике на основании данных по безопасности, которые:
а) оказали существенное влияние на соотношение польза-риск зарегистрированного лекарственного препарата; и/или
б) оказали влияние на проведение конкретного клинического исследования(-ий) или в целом на программу клинической разработки лекарственного препарата.
В разделе должны быть указаны основания для принятия данных мер и при необходимости дополнительная информация, если таковые доступны.
Меры, принятые в отношении исследуемого лекарственного препарата могут включать:
а) отказ в выдаче разрешения на проведение клинического исследования/испытания по аспектам безопасности или этическим вопросам;
б) частичная или полная приостановка клинического исследования/испытания либо полная остановка клиническая исследования/испытания ранее планируемого срока по причине выявленных данных по безопасности или недостаточной терапевтической эффективности;
в) отзыв исследуемого лекарственного препарата или препарата сравнения;
г) отказ в получении разрешения на применение по показанию, исследуемому в ходе клинического исследования, включая добровольный отзыв подачи заявления на регистрацию;
д) введение мер минимизации риска, включая:
— изменения в протокол исследования/испытания, обусловленные данными по безопасности или эффективности (такие, как изменение режима дозирования, изменения критериев включения/невключения, введение дополнительных мер по мониторингу субъектов исследования, ограничение продолжительности исследования/испытания);
— ограничения исследуемой популяции или показаний к применению:
— изменения информированного согласия, связанные с аспектами профиля безопасности;
— изменения состава;
— дополнительное требование регуляторных органов по особому порядку представления информации по безопасности лекарственного препарата;
— специальное информирование врачей-исследователей или медицинских работников; и
— планирование проведения новых исследований по оценке аспектов профиля безопасности.
Меры, принятые в отношении зарегистрированного лекарственного препарата включают:
а) отказ в продлении действия регистрационного удостоверения;
б) приостановка или отзыв регистрационного удостоверения;
в) введение плана минимизации риска, включая:
— существенные ограничения в распространении или введение иных мер минимизации риска;
— существенные изменения инструкции по медицинскому применению, которые могут повлиять на программу разработки, включая ограничения показаний к назначению или групп пациентов, которым назначается лекарственный препарат;
— специальное информирование медицинских работников; и
— требование по проведению пострегистрационного исследования со стороны регуляторных органов.
2.2.4 Раздел ПОБ «Изменения, внесенные в справочную информацию по безопасности лекарственного препарата»
В разделе перечисляется информация обо всех существенных изменениях, внесенных в справочную информацию по безопасности лекарственного препарата за отчетный период. Данные существенные изменения включают изменения в разделы противопоказаний, предостережений, особых указаний, дополнение информацией о серьезных нежелательных реакциях, нежелательных реакциях, представляющих особый интерес, реакциях взаимодействия; важные данные продолжающихся и завершенных клинических исследований/испытаний; важные данные доклинических исследований (например, изучение канцерогенности). Информация по данным изменениям должна быть представлена в соответствующих разделах ПОБ. В приложении к ПОБ должна прилагаться версия справочной информации по безопасности лекарственного препарата с соответствующими изменениями.
Держатель регистрационного удостоверения также представляет информацию о внесенных и находящихся на этапе внесения изменениях в инструкцию по медицинскому применению на основании обновленной версии основной информации по безопасности держателя регистрационного удостоверения с представлением ее в приложении.
2.2.5. Раздел ПОБ «Оценка количества пациентов, подвергшихся воздействию»
ПОБ должен содержать точную оценку количества пациентов, которые подверглись воздействию лекарственного препарата, включая все данные в отношении объема продаж и количества назначений. Данная оценка должна сопровождаться качественным и количественным анализом применения в реальной медицинской практике с указанием того, каким образом это может отличаться от одобренного применения, основываясь на всех данных доступных держателю регистрационного удостоверения и результатах наблюдательных исследований по оценке использования лекарственного препарата.
В данном разделе должна быть представлена оценка объема и характеристики популяции, подвергшейся воздействию лекарственного препарата, включая краткое описание используемого для оценки метода и указания недостатков используемого метода.
Согласующиеся методы по оценке воздействия на субъекта/пациента должны использоваться во всех разделах ПОБ для одного лекарственного препарата. Если уместным является замена используемого метода оценки, оба метода и расчеты по ним должны быть представлены в ПОБ с объяснением замены.
2.2.5.1. Подраздел ПОБ «Общее количество пациентов, подвергшихся воздействию в клинических исследованиях»
Данный раздел ПОБ должен содержать следующую информацию по пациентам, включенным в клинические исследования/испытания (рекомендуется табличный формат):
а) кумулятивное число субъектов исследования, включенных в продолжающиеся и завершенные клинические исследования/испытания и подвергшихся воздействию исследуемого лекарственного препарата, плацебо, или/и активного препарата сравнения от международной даты одобрения разрабатываемого лекарственного препарата. Может признаваться, что для лекарственных препаратов, длительное время находящихся в обращении, детальная информация может не быть доступна;
б) более детальная кумулятивная информация по субъектам исследования, подвергшимся воздействию, при наличии (например, сгруппированная по возрасту, полу, расовой принадлежности по всей программе разработки);
в) важные различия между исследованиями/испытаниями в отношении назначаемых доз, путей введения, подгрупп пациентов;
г) в случае, если клинические исследования/испытания проводились на особых группах пациентов (например, беременные женщины, пациенты с нарушениями функции почек, печени, сердечно-сосудистой системы; пациенты с клинически значимым генетическим полиморфизмом), должны быть представлены данные по воздействию;
д) при наличии существенных различий по времени воздействия между субъектами, рандомизированными на получение исследуемого лекарственного препарата или препарата(-ов) сравнения, или несоответствий по продолжительности воздействия между клиническими исследованиями/испытаниями, необходимо сделать оценку воздействия в выражении субъект-время (пациенто-дни, -месяцы или –годы);
е) данные по воздействию исследуемого препарата на здоровых добровольцев может иметь меньшую значимость для оценки профиля безопасности лекарственного средства в целом, в зависимости от типа наблюдаемых нежелательных реакций, в особенности, когда пациенты подвергаются воздействию единичной дозы. Подобные данные должны быть представлены отдельно с пояснениями в случае необходимости;
ж) в случае, если в обобщенной информации по нежелательным реакциям, выявленным в ходе клинических исследований/испытаний, указаны серьезные нежелательные реакции, должно быть сделано соответствующее указание по оценке воздействия на пациента, когда это возможно;
з) для определенных особо важных клинических исследований/испытаний демографическая характеристика пациентов должна быть представлена отдельно.
2.2.5.2. Подраздел ПОБ «Общее количество пациентов, подвергшихся воздействию, по данным применения на рынке»
В случаях, когда это возможно, должна быть представлена отдельная оценка по кумулятивному воздействию (начиная от международной даты регистрации) и воздействию за определенный интервал (от даты окончания сбора данных по предшествующему ПОБ). В разделе должна быть представлена оценка по количеству пациентов, подвергшихся воздействию и методу(-ам), с помощью которых выполнялось определение и оценка. Должно быть представлено обоснование, если выполнение расчета числа пациентов, подвергшихся воздействию, невозможно осуществить. Если невозможно выполнить оценку числа пациентов, должны быть представлены альтернативные варианты оценки с указанием метода (-ов) их выполнения. Примером альтернативного показателя оценки воздействия является показатель пациенто-дней и число назначений (выписываний). Только в тех случаях, когда данные показатели недоступны, может быть использована оценка объема продаж, выраженная в весовых единицах или дозах. Может быть применена концепция установленной суточной дозы (DDD)для получения данных по воздействию на пациентов.
Данные по воздействию должны быть приведены по следующим категориям использования лекарственного препарата:
- Пострегистрационное применение (за исключением клинических исследований/испытаний):
Должна быть представлена общая оценка. В дополнение данные должны быть представлены с разбивкой по полу, возрасту, показаниям, дозам, формам выпуска и регионам, где это применимо. В зависимости от лекарственного препарата, иные переменные могут быть приведены, как значимые, например число выполненных вакцинаций, способ введения и продолжительность лечения.
В случае, если была выявлена серия сообщений о нежелательных реакциях, предполагающая наличие сигнала, должны быть представлены данные по воздействию внутри соответствующей подгруппы, если это возможно.
- Пострегистрационное применение у особых популяционных групп:
В случае, если на пострегистрационном этапе лекарственный препарат используется у особых популяционных групп, должна быть представлена доступная информация в отношении кумулятивного числа пациентов, подвергшихся воздействию, и используемый метод расчета. Источники этих данных могут включать неинтервенционные исследования, разработанные непосредственно для получения данных по особым популяционным подгруппам, включая регистры. Популяции, включаемые в оценку по данному разделу включают, но не ограничиваются следующими:
— педиатрическая популяция;
— популяция пожилого возраста;
— женщины в период беременности и кормления;
— пациенты с нарушениями функции печени и/или почек;
— пациенты с иной важной сопутствующей патологией;
— пациенты, степень тяжести заболевания которых отлична от исследуемой в ходе клинических исследований;
— подпопуляции с носительством генетического полиморфизма (-ов);
— пациенты с иной расовой или этнической принадлежностью.
- Особенности применения лекарственного препарата
В случае, если держателю регистрационного удостоверения становится известна информация об определенных особенностях применения лекарственного препарата, должно быть приведено описание данных особенностей и сделана соответствующая оценка и интерпретация данных по безопасности. К числу таких особенностей относится, в частности, применение в медицинской практике по показаниям, не включенным в число одобренных. Если имеются соответствующие данные, держатель регистрационного удостоверения может сделать комментарий в отношении того, насколько данное применение поддерживается клиническими протоколами, доказательной базой клинических исследований, либо обусловлено отсутствием в целом зарегистрированных альтернатив. Должна быть представлена количественная оценка по объему данного применения, если подобные данные имеются.
2.2.6. Раздел ПОБ «Обобщенные табличные данные»
Целью данного раздела ПОБ является представление данных по нежелательным реакциям/явлениям, выявленным в ходе клинических исследований в форме обобщенных табличных данных. На усмотрение держателя регистрационного удостоверения может быть приведено графическое отображение определенных аспектов данных с целью облегчения восприятия и понимания.
Отнесение к числу серьезных нежелательных реакций в обобщенных табличных данных должно соответствовать отнесению, сделанному по результатам оценки индивидуальных сообщений о нежелательных реакциях с использованием критериев серьезности, установленных законодательством. Оценка серьезности не должна изменяться при подготовке данных для включения в ПОБ.
2.2.6.1 Подраздел ПОБ «Справочная информация»
В данном подразделе указывается версия терминологического классификатора, используемого для анализа нежелательных явлений/реакций.
2.2.6.2 Подраздел ПОБ «Обобщенные табличные данные по серьезным явлениям, выявленным в ходе клинических исследований/испытаний»
В данном подразделе ПОБ должно быть приведено обоснование по приложению, которое включает кумулятивные обобщенные табличные данные по серьезным нежелательным явлениям, которые были выявлены в ходе клинических исследований/испытаний, организованных держателем регистрационного удостоверения, начиная от международной даты одобрения разрабатываемого лекарственного препарата до даты окончания сбора данных по текущему ПОБ. Держателем регистрационного удостоверения должны быть сделаны объяснения всех исключаемых данных (например, данные по результатам клинического исследования/испытания могут быть недоступны на протяжении нескольких лет). Данные в табличной форме должны быть сгруппированы в соответствии с классификационным отнесением нежелательных реакций по органо-системным классам для исследуемого лекарственного препарата, а также для препаратов сравнения (активных и плацебо). Когда это является целесообразным, данные должны быть представлены в группированном виде по клиническим исследованиям/испытаниям, показаниям, путям введения и иным переменным.
Следующие аспекты должны быть рассмотрены:
а) Рекомендуется представление оценки причинно-следственной связи по редким нежелательным реакциям. Следует представлять данные по всем серьезным нежелательным явлением и для исследуемого лекарственного препарата, и для препаратов сравнения и плацебо с тем, чтобы была возможность сделать групповое сравнение, в том числе, в отношении частоты. Полезным является представление данных с отражением взаимосвязи назначаемой дозы и частоты.
б) Обобщенные табличные данные должны включать как ослепленные, так и разослепленные данные по серьезным нежелательным явлениям в клинических исследованиях. Разослепленные данные могут быть представлены по результатам завершенных клинических исследований и отдельным индивидуальным случаям, которые были разослеплены по определенным причинам, например по аспектам безопасности или выполнения требований незамедлительного репортирования. Спонсоры/заявители клинического исследования/испытания и держатели регистрационных удостоверений не выполняют разослепление непосредственно в связи с подготовкой ПОБ.
в) Определенные нежелательные реакции могут быть исключены из обобщающей информации, но все подобные исключения должны обосновываться в отчете. Например, нежелательные реакции, которые были определены в протоколе как исключающиеся из процедуры незамедлительного репортирования и только включаемые в общую базу данных по причине того, что они являются присущими целевой популяции, или совпадают с конечными точками.
2.2.6.3. Подраздел ПОБ «Обобщенные табличные данные по данным пострегистрационного применения»
В данном разделе ПОБ представляется обоснование по приложению, включающему в табличной форме обобщающие данные по нежелательным реакциям кумулятивно за весь период и за отчетный период, от даты международной регистрации лекарственного препарата до даты окончания сбора данных, Включаются сведения о нежелательных реакциях, полученных в ходе неинтервенционных исследований и спонтанного репортирования, включая данные от медицинских и фармацевтических работников, потребителей, пациентов, регуляторных органов и данных, опубликованных в медицинской литературе. Серьезные и несерьезные нежелательные реакции должны быть представлены в отдельных таблицах. В таблице данные должны быть распределены согласно классификации по органо-функциональным классам. По особо важным аспектам профиля безопасности могут быть представлены отдельные таблицы нежелательных реакций с группированием данных по показаниям, способу введения и иным параметрам.
2.2.7 Раздел ПОБ «Резюме важных данных, полученных в ходе клинических исследований/испытаний за отчетный период»
Держатель регистрационного удостоверения должен представить в приложении перечисление организованных им интервенционных клинических исследований с целью возможности идентификации, характеристики и количественной оценки уровня рисков, подтверждения профиля безопасности лекарственного препарата или оценки эффективности мер минимизации риска, которые были завершены или продолжают выполняться в отчетный период.
Если это представляется возможным, данные следует разбить на категории по половому и возрастному признаку (в особенности взрослые по сравнению с детской популяцией), показаниям, режимам дозирования и регионам.
Сигналы, выявленные в ходе клинических исследований, должны быть представлены в табличной форме в разделе 15 ПОБ («Обзор по сигналам: новые, находящиеся в работе или завершенные»). Для сигналов делается оценка, являются ли данные сигналы потенциальными или идентифицированными рисками, риск должен быть оценен и характеризован в разделе 16.3 ПОБ («Оценка рисков и новой информации») и 16.4 («Характеристика рисков»), соответственно.
В данном разделе ПОБ должна быть представлена обобщающая информация по клинически важным данным по эффективности и безопасности, полученным из следующих источников за отчетный период:
2.2.7.1. Подраздел «Завершенные клинические исследования» Данный подраздел ПОБ должен представлять краткую информацию по клинически важным данным по эффективности и безопасности, полученным в результате завершенных за отчетный период клинических исследований/испытаний. Данная информация должна быть представлена в сжатом виде или в форме синопсиса. Он может включать информацию, которая подтверждает или опровергает ранее идентифицированные сигналы по безопасности, а также доказательства по новым сигналам по безопасности.
2.2.7.2. Подраздел ПОБ «Продолжающиеся клинические исследования/испытания»
В случае, если держателю регистрационного удостоверения становится известна какая-либо клинически важная информация, которая была получена в ходе продолжающихся клинических исследований/испытаний (например, выявленная в ходе промежуточного анализа безопасности либо в результате разослепления выявленных серьезных нежелательных явлений), в данном разделе должна быть кратко изложена информация по выявленной новой информации по безопасности. Этот раздел также может включать информацию, которая подтверждает или опровергает ранее идентифицированные сигналы по безопасности, а также доказательства по новым сигналам по безопасности.
2.2.7.3 Подраздел ПОБ «Длительное последующее наблюдение»
В тех случаях, когда имеются данные по длительному последующему наблюдению пациентов, включенных в клинические исследования/испытания, в разделе приводится информация по полученным в результате длительного последующего наблюдения значимым с точки зрения профиля безопасности данным.
2.2.7.4. Подраздел ПОБ «Иное терапевтическое применение лекарственных препаратов»
Данный подраздел ПОБ должен включать клинически важную информацию по безопасности, полученную в результате других программ, проведенных держателем регистрационного удостоверения, по специальным протоколам (например, программы расширенного доступа, программы использования в связи с исключительными обстоятельствами из соображений сострадания, индивидуального доступа и иные).
2.2.7.5. Подраздел ПОБ «Новые данные по безопасности в отношении назначения фиксированных комбинаций»
В случае, если иное не определено национальными регуляторными органами, следующие данные должны быть представлены в отношении комбинированной терапии:
а) В случае, если лекарственный препарат также одобрен для назначения в качестве компонента фиксированной лекарственной терапии или многокомпонентного режима лекарственной терапии, в разделе должны быть обобщены важные данные по безопасности применения комбинированной терапии.
б) В случае, если лекарственный препарат является комбинированным лекарственным препаратом, данный раздел должен обобщать важную информацию по безопасности по каждому из индивидуальных компонентов.
2.2.8. Раздел ПОБ «Данные неинтервенционных исследований»
В данном разделе обобщается соответствующая информация по безопасности или данные об их влиянии на оценку соотношения польза-риск, полученная по результатам неинтервенционных клинических исследований/испытаний (например, обсервационных исследований, эпидемиологических исследований, регистров, программ активного мониторинга), организованных держателем регистрационного удостоверения, которая стала доступна в отчетный период. Раздел должен включать данные, имеющие отношение к аспектам профиля безопасности, полученные по результатам исследований по оценке использования лекарственного препарата.
Держатель регистрационного удостоверения должен включать в приложение к отчету перечень всех неинтервенционных исследований/испытаний, организованных держателем регистрационного удостоверения, выполненных с целью выявления, характеристики и количественной оценки вызывающих опасения аспектов профиля безопасности, подтверждения профиля безопасности лекарственного препарата или оценки эффективности мер минимизации риска, которые были выполнены или выполняются на протяжении отчетного периода (например, пострегистрационные исследования безопасности).
Отчеты о стадии выполнения или итоговые отчеты, которые были подготовлены на протяжении отчетного периода, должны включаться в приложение к ПОБ.
2.2.9. Раздел ПОБ «Данные других клинических исследований и из других источников»
В разделе должна быть обобщена информация, имеющая отношение к оценке соотношения польза-риск лекарственного препарата и полученная по результатам иных клинических исследований либо полученная из иных источников, к которым имелся доступ у держателя регистрационного удостоверения, за отчетный период (например, результаты мета-анализов рандомизированных клинических исследований, данные по безопасности партнеров по разработке лекарственного препарата и иные).
2.2.10. Раздел ПОБ «Данные доклинических исследований»
В разделе представляется обобщенная информация по значимым в отношении профиля безопасности данным, полученным в результате доклинических исследований in vivo и in vitro (например, исследования канцерогенности, репродуктивной токсичности или иммунотоксичности), выполняемых или завершенных в отчетный период. Оценка влияние полученных данных на профиль безопасности должна быть представлена в разделе 16 («Сигнал и оценка риска») и разделе 18 («Интегрированный анализ соотношения польза-риск по одобренным показаниям») ПОБ.
2.2.11. Раздел ПОБ «Литература»
Раздел включает обобщение полученных новых и значимых данных по безопасности, которые были опубликованы в прошедшей экспертную оценку научной литературе, либо были получены из неопубликованных монографий, которые имеют отношение к лекарственному препарату и стали доступны держателю регистрационного удостоверения в отчетный период.
Литературный поиск для подготовки ПОБ должен быть шире, чем осуществляемый с целью поиска индивидуальных сообщений о нежелательных реакциях, поскольку должен включать также исследования, в ходе которых были оценены исходы с точки зрения безопасности в группах субъектов исследования.
Особые аспекты профиля безопасности, которые должны включаться в поиск, но которые могут не быть выявлены при его осуществлении с целью получения данных по индивидуальным случаям нежелательных реакций, включают:
а) исходы беременности (включая прерывание), которые не сопровождаются нежелательными последствиями;
б) применение в педиатрической популяции;
в) применение по программам использования в связи с исключительными обстоятельствами из соображений сострадания, персонализированным программам назначения;
г) отсутствие эффективности;
д) асимптоматическая передозировка, несоответствующее и неправильное применение;
е) медицинские ошибки, не сопровождавшиеся развитием нежелательных явлений;
ж) важные результаты доклинических исследований.
В случае, если это применимо, в разделе должна быть рассмотрена информация по другим активным веществам данной группы.
2.2.12 Раздел ПОБ «Другие периодические отчеты»
Данный раздел ПОБ применим только в тех определенных случаях, когда по договоренности с регуляторными органами держателем регистрационного удостоверения готовится более одного ПОБ на лекарственный препарат: в случае фиксированной комбинации, лекарственного препарата с множественными показаниями и/или формами выпуска. В целом держатель регистрационного удостоверения готовит один ПОБ на одно действующее вещество (за исключением случает, если иное не требуется регуляторными органами); в случае если подготовлено несколько ПОБ для одного лекарственного препарата, в данном разделе должны быть обобщены значимые данные по безопасности из других ПОБ, если она не представлена в иных разделах данного ПОБ.
В случае наличия доступа, на основании контрактных договоренностей, держатель регистрационного удостоверения должен представить в обобщенном виде значимые данные по безопасности, полученные в представляемых в ПОБ за отчетный период другими сторонами (например, спонсорами или иными партнерами).
2.2.13. Раздел ПОБ «Недостаточная терапевтическая эффективность в контролируемых клинических исследованиях»
Данные, полученные в ходе клинических исследований, которые могут свидетельствовать о недостаточной терапевтической эффективности, либо недостаточной терапевтической эффективности в отношении принятой терапии, для лекарственных препаратов, используемых для лечения и профилактики серьезных и жизнеугрожающих заболеваний, эти данные могут свидетельствовать о значительном риске для целевой популяции и должны быть проанализированы и обобщены в данном разделе ПОБ.
В случае, если это применимо к оценке соотношения польза-риск, данные клинических исследований, демонстрирующие недостаточную терапевтическую лекарственных препаратов, не предназначенных для лечения жизнеугрожающей патологии, должны также быть проанализированы в данном разделе.
2.2.14. Раздел ПОБ «Важная информация, полученная после завершения подготовки ПОБ»
В разделе обобщаются потенциально важные данные по безопасности и эффективности, которые были получены после даты окончания сбора данных, но в период подготовки ПОБ. Примеры включают данные клинически значимых новых публикаций, важные данные последующего наблюдения, клинически важные токсикологические данные, а также все действия держателя регистрационного удостоверения, комитетов по оценке данных, регуляторных органов, предпринятые в связи с аспектами профиля безопасности. Новые индивидуальные сообщения о нежелательных реакциях не должны включаться в раздел, за исключением случаев, когда они могут представлять важный показательный случай (например, первый случай развития важного нежелательного явления) или важный сигнал по безопасности.
Данные раздела должны быть учтены при оценке риска и новой информации.
2.2.15. Раздел ПОБ «Обзор сигналов: новые, рассматриваемые и завершенные»
Целью данного раздела является представление исчерпывающего обзора выявленных сигналов, сигналов на этапе оценки и прошедших оценку за отчетный период.
Держателем регистрационного удостоверения должно быть представлено краткое описание метода, который используется для выявления сигналов, а также источники данных для выявления сигналов.
К новым выявленным сигналам относятся сигналы, которые были выявлены за отчетный период. К рассматриваемым сигналам относят сигналы, которые находились на этапе оценки на дату окончания сбора данных. К завершенным относят сигналы, оценка которых была завершена за отчетный период. Сигналы, которые являются одновременно и новыми и завершенными за отчетный период должны быть отнесены в раздел завершенных сигналов.
Раздел должен включать данные по рассматриваемым и завершенным за отчетный период сигналам, приведенные в табличной форме. Таблица прилагается к отчету в форме приложения. На усмотрение держателя регистрационного удостоверения, данная информация может также включать кумулятивные данные по сигналам, включая ранее завершенные сигналы, при этом должна быть указана дата, от которой выполнено обобщение по сигналам.
Детальная оценка сигналов включается в раздел ПОБ 16.2 («Оценка сигнала») и 16.3 («Оценка рисков и новой информации»).
2.2.16. Раздел ПОБ «Сигналы и оценка риска»
2.2.16.1. Подраздел ПОБ «Обобщенная информация по проблемам по безопасности»
Целью подраздела является представление базисной обобщающей информации по важным аспектам профиля безопасности, представляющим проблемы по безопасности, с указанием по каждой проблеме по безопасности какая новая информация и оценка по данным аспектам может быть сделана. При определении важности каждого из аспектов риска следует рассмотреть следующие факторы:
а) серьезность риска с медицинской точки зрения, включая влияние на индивидуальное состояние пациентов;
б) частота, предсказуемость, предотвратимость и обратимость;
в) потенциальное влияние на общественное здоровье (частота в популяции; размер популяции, подвергшийся воздействию); и
г) общественное приятие риска в случаях возможности влияния на общественное здоровье (например, отказ от программы вакцинации).
Обобщающая информация должна представлять имеющиеся сведения по лекарственному препарату от начала отчетного периода ПОБ и отражать:
а) важные идентифицированные риски;
б) важные потенциальные риски;
в) важную отсутствующую информацию.
Для лекарственных препаратов, имеющих спецификацию по безопасности, информация, включаемая а данный подраздел, должна совпадать с обобщающей информацией, представленной в текущей версии спецификации по безопасности на момент начала отчетного периода ПОБ.
Для лекарственных препаратов, не имеющих спецификацию по безопасности, данный подраздел должен представлять информацию по важным идентифицированным, потенциальным рискам и важной отсутствующей информации, связанной с применением лекарственного препарата на основании данных до- и пострегистрационного периода. Примеры могут включать следующую информацию:
а) важные нежелательные реакции;
б) взаимодействия с другими лекарственными препаратами;
в) выявленные медицинские ошибки или промахи, в случаях, не сопровождавшихся развитием нежелательных реакций;
г) взаимодействия с продуктами питания или иными веществами;
д) результаты воздействия при выполнении профессиональной деятельности;
е) классовые фармакологические эффекты.
Обобщение по важной отсутствующей информации должно оценить критичность пробелов в имеющихся знаниях по определенным аспектам профиля безопасности для целевых популяций.
2.2.16.2. Подраздел ПОБ «Оценка сигнала»
Информация, представляемая в подразделе, должна обобщать результаты оценки сигналов по безопасности, которая была завершена в отчетный период; могут быть две основных категории:
- Сигналы, которые по результатам оценки могут быть отнесены к категории потенциальных или идентифицируемых рисков, включая отсутствие терапевтической эффективности. Данные завершенные сигналы обсуждаются в разделе 16.3 ПОБ («Оценка рисков и новой информации»).
- Сигналы, которые по результатам оценки были отклонены как ложные сигналы на основании научной оценки имеющейся на момент проведения процедуры информации. По данной категории сигналов должно быть представлено описание каждого сигнала с целью обоснования его отклонения от категории сигнала. Данное описание может быть включено в основной текст ПОБ или в приложение к отчету.
Для сигналов, по которым была завершена процедура оценки за отчетный период, рекомендуется соблюдение соответствия между объемом и детализацией данных по оценке сигнала и значимостью данного аспекта профиля безопасности для общественного здоровья, а также степенью достаточности доказательной базы. Данная информация должна включать следующие аспекты:
а) источник или побудительный момент формирования сигнала;
б) обоснование, имеющее отношение к оценке;
в) методы оценки, включая источники данных, критерии поиска или аналитические подходы;
г) результаты: обобщенная информация по критическому анализу данных, рассматриваемых при оценке сигнала;
д) обсуждение;
е) заключение, включая предлагаемые действия.
2.2.16.3. Подраздел ПОБ «Оценка рисков и новой информации»
Держатель регистрационного удостоверения должен представить критическую оценку новой информации за отчетный период в отношении новых или ранее выявленных рисков (важную или иную).
Данный подраздел ПОБ должен содержать описание и оценку всех рисков, выявленных за отчетный период, а также оценку влияния новых данных на ранее выявленные риски. В раздел не включается обобщающая или повторяющаяся информация, включенная в другие разделы ПОБ, но представляется интерпретация и оценка новой информации применительно к характеристике профиля риска.
Новая информация должна быть представлена по следующим разделам:
- новые потенциальные риски;
- новые идентифицированные риски;
- новая информация по ранее выявленным рискам (потенциальным и идентифицированным);
- обновление по важной отсутствующей информации.
Представляется краткое описание важных рисков. Для рисков, относимых к иным и не относимых к «важным» рискам, по которым была получена новая информация за отчетный период, уровень детализации должен соответствовать имеющейся доказательной базе по данному риску и значимости его влияния на общественное здоровье.
Вся новая информация по влиянию лекарственного препарата на популяцию либо данные по ранее отсутствующей информации должны быть критически оценены. Указывается, какие из аспектов профилей безопасности, вызывающих опасения, и неясных аспектов профиля безопасности, остались невыясненными.
2.2.16.4. Подраздел ПОБ «Характеристика рисков»
В подразделе дается характеристика важных идентифицированных рисков и важных потенциальных рисков на основании кумулятивных данных (в том числе не ограничиваемых отчетным периодом) и описывается важная отсутствующая информация.
В случае, когда это применимо, принимая во внимание источник данных, информация по рискам должна включать следующее:
а) частота;
б) число выявленных случаев (нумератор); точность оценки, принимая во внимание источник данных;
в) объем назначений (деноминатор), выраженное как число пациентов, пациенто-месяцев (-лет) и т.д.; точность оценки;
г) оценка относительного риска и точность оценки;
д) оценка абсолютного риска и точность оценки;
е) влияние на пациента (влияние на симптомы, качество жизни);
ж) влияние на общественное здоровье;
з) факторы риска (например, индивидуальные факторы риска (рассматривается возраст, беременность/лактация, нарушение функции печени/почек, значимая сопутствующая патология, степень тяжести заболеваний, генетический полиморфизм, расовая и/или этническая принадлежность), доза);
и) продолжительность лечения, период риска;
к) предотвратимость (оценивается предсказуемость, возможность мониторировать состояние по индикаторным симптомам или лабораторным параметрам);
л) обратимость;
м) потенциальный механизм;
н) уровень доказательности и неопределенности, включая анализ противоречащих фактов при их наличии.
При подготовке ПОБ для лекарственных препаратов с несколькими показаниями, формами выпуска или способами введения, в случае наличия существенных различий по идентифицированным и потенциальным рискам, может быть обоснованным представление данных по рискам раздельно по показаниям, формам выпуска или способам введения. Могут быть представлены следующие разделы:
а) риски, характерные для действующего вещества;
б) риски, характерные для определенных форм выпуска или способов введения (включая воздействие при выполнении профессиональной деятельности);
в) риски, характерные для определенных популяций;
г) риски, связанные с применением без назначения врача (для действующих веществ, которые представлены в формах, отпускаемых по рецепту и без рецепта);
д) проблемы по безопасности, связанные с отсутствующей информацией.
2.2.16.5. Подраздел ПОБ «Эффективность мер минимизации риска (если применимо)»
Меры минимизации риска включают в себя действия, направленные на предотвращение нежелательных реакций, связанных с воздействием лекарственного препарата, либо снижение степени их тяжести при возникновении. Целью деятельности по минимизации риска является снижение вероятности развития или степени тяжести нежелательных лекарственных реакций. Меры минимизации риска включают рутинные меры минимизации риска (например, изменения в инструкцию по медицинскому применению) или дополнительные меры минимизации риска (например, прямое информирование специалистов системы здравоохранении/образовательные материалы).
В подразделе должны быть представлены результаты оценки эффективности мер минимизации риска. В обобщенном виде представляется соответствующая информация по эффективности и/или ограничениям конкретных мер минимизации риска по важным идентифицированным рискам, которая была получена за отчетный период. Результаты оценки за отчетный период представляются в приложении к отчету.
2.2.17. Раздел ПОБ «Оценка пользы»
2.2.17.1 Подраздел ПОБ «Важная базисная информация по эффективности в ходе клинических испытаний и применения в медицинской практике»
В подразделе суммируется основная информация по эффективности лекарственного препарата в ходе клинических исследований и эффективность, продемонстрированная при применении в медицинской практике от начала отчетного периода. Данная информация должна иметь отношение к одобренным показаниям к применению.
Для лекарственных препаратов с несколькими показаниями, целевыми популяциями и/или способами введения польза должна быть охарактеризована в отдельности по каждому фактору.
Для лекарственных препаратов, у которых за отчетный период были выявлены существенные изменения профиля безопасности или эффективности, данный подраздел должен включать достаточную информацию по обоснованию обновленной характеристики пользы лекарственного препарата, отраженной в подразделе ПОБ 17.3 («Характеристика пользы»). Содержание и степень детализации информации, представленной в разделе, может варьироваться по различным лекарственным препаратам, включая в случаях, когда это применимо, следующие аспекты:
а) эпидемиология и происхождение заболевания;
б) характеристика пользы (например, диагностическое, профилактическое, симптоматическое, болезнь-модифицирующее);
в) важные конечные точки, подтверждающие пользу (например, влияние на смертность, симптоматику, исходы);
г) доказательства эффективности в клинических исследованиях и медицинской практике по сравнению с препаратом сравнения (например, сравнительные клинические исследования с активным контролем, мета-анализы, обсервационные исследования); и
д) тенденции и/или доказательства пользы по важным популяционным подгруппам (например, возрастным, половым, этническим, по степени тяжести заболевания, генетическому полиморфизму) в случае, если это имеет отношении к оценке соотношения польза-риск.
2.2.17.2 Подраздел ПОБ «Новая выявленная информация по эффективности в ходе клинических исследований и применения в медицинской практике»
Для некоторых лекарственных препаратов за отчетный период может быть получена новая информация по эффективности в клинических исследованиях и медицинской практике, которая должна быть представлена в подразделе. Отдельная информация по доказательной базе в отношении не одобренных показаний применению не включается в раздел, за исключением случаев, когда это имеет отношение к оценке соотношения польза-риск.
Особое внимание в подразделе уделяется вакцинам, антиинфекционным и иным препаратам, для которых изменения терапевтической среды могут повлиять на соотношение польза/риск с течением времени.
Содержание и степень детализации информации, представляемой в данном разделе, может варьироваться в зависимости от лекарственного препарата; в случае отсутствия новой информации за отчетный период, может быть сделана ссылка на раздел 17.1 («Важная базисная информация по эффективности в клинических исследованиях и медицинской практике»).
2.2.17.3 Подраздел ПОБ «Характеристика пользы»
В подразделе представляется объединенная информация по базисным и новым данным по терапевтической пользе, которые стали известны за отчетный период по одобренным показаниям.
В случае отсутствия новых данных по профилю пользы и отсутствия значительных изменений профиля безопасности, данный подраздел должен содержать ссылку на подраздел 17.1 («Важная базисная эффективность в клинических исследованиях и информация по эффективности в медицинской практике»).
В случае если за отчетный период была получена новая информация по терапевтической пользе и не было значимых изменений профиля безопасности, в разделе кратко приводятся объединенные данные по базисной и новой информации.
В случае наличия существенных изменений профиля безопасности, либо получения новых данных, предполагающих значительно меньший уровень терапевтической пользы по сравнению с изначально продемонстрированным, в разделе должна быть приведена краткая, но критическая оценка доказательной базы по безопасности и эффективности в ходе клинических исследований и медицинской практике, с приведением данных по следующим аспектам:
а) краткое описание доказательного уровня данных по терапевтической пользе; рассматривается сравнительный аспект эффективности, степень выраженности эффекта, правильность статистической обработки, слабые и сильные аспекты методологии, соответствие данных в разных исследованиях/испытаниях;
б) новая информация, поставившая под сомнение суррогатные конечные точки, если таковые использовались;
в) клиническая значимость выраженности терапевтического эффекта;
г) обобщаемость терапевтического эффекта между целевыми подгруппами (например, информация о недостаточности терапевтического эффекта по какой-либо популяционной подгруппе);
д) адекватность характеристики доза-терапевтический ответ;
е) продолжительность эффекта;
ж) сравнительная эффективность; и
з) определение степени, в которой данные по эффективности, полученные в клинических исследованиях, могут быть обобщены с популяцией, в которой применяется лекарственный препарат в медицинской практике.
2.2.18. Раздел ПОБ «Интегрированный анализ соотношения польза-риск по одобренным показаниям»
В разделе держателем регистрационного удостоверения должна быть представлена обобщенная оценка пользы и риска лекарственного препарата при его применении в клинической практике. Представляется критический анализ и объединенная информация по предыдущим разделам в части пользы и риска без дублирования с информацией в разделах 16.3 («Оценка рисков и новой информации») и 17.3 («Характеристика пользы»).
8.5.18.1. Подраздел ПОБ «Контекст соотношения польза-риск – медицинская потребность в лекарственном препарате и важные альтернативы»
В подразделе представляется краткое описание медицинской потребности в лекарственном препарате по одобренным показаниям и суммировано по альтернативам (медикаментозным, хирургическим или иным; включая отсутствие лечения).
2.2.18.2. Подраздел ПОБ «Оценка процедуры анализа соотношения польза-риск»
Соотношение польза-риск имеет различное значение в зависимости от показаний и целевых популяций. Следовательно, для лекарственных препаратов, зарегистрированных по нескольким показаниям, соотношение польза-риск должно быть оценено отдельно по каждому показанию. В случае наличия существенных различий соотношения польза-риск между подгруппами в рамках одного показания, оценка соотношения польза-риск должна быть представлена отдельно и для популяционных подгрупп, если это возможно.
а) Основные вопросы в отношении пользы и рисков:
Ключевая информация, представленная в предшествующих разделах по пользе и риску, должна быть объединена с целью оценки их соотношения.
Оценивается контекст применения лекарственного препарата: излечение, профилактика, диагностика; степень тяжести и серьезность заболевания; целевая популяция (относительно здоровые, хронические заболевания).
В отношении пользы оценивается ее характер, клиническая значимость, продолжительность эффекта, обобщаемость, доказательство эффективности у пациентов, не отвечающих на альтернативное лечение, выраженность эффекта, индивидуальные элементы пользы.
В отношении риска оценивается клиническая значимость (например, характер токсичности, серьезность, частота, предсказуемость, предотвратимость, обратимость, влияние на пациента), а также аспекты риска, связанные с применением не по одобренным показаниям, новым показаниям, неправильным применением.
При формулировке оценки соотношения польза-риск рассматриваются слабые и сильные стороны, а также неопределенности доказательной базы с описанием их влияния на оценку. Приводится характеристика ограничений выполненной оценки.
б) Представляется описание и аргументации используемой методологии для оценки соотношения польза-риск:
предположения, рассмотрение, соотнесения, которые подтверждают сделанный вывод по оценке соотношения польза-риск;
комментарии в отношении возможности выражения пользы и риска в представленном виде и их сопоставления;
если представлена количественная оценка соотношения, включается обобщенное описание методов оценки;
экономическая оценка (например, стоимость-эффективность) не должна рассматриваться при оценке соотношения польза-риск.
2.2.19. Раздел ПОБ «Заключение и действия»
Заключительный раздел ПОБ должен содержать заключение о влиянии всей новой информации, выявленной в отчетный период, на общую оценку соотношения польза-риск по каждому одобренному показанию, а также подгруппам пациентам, в случаях, когда это применимо.
Основываясь на оценке кумулятивных данных по безопасности и анализу соотношения польза-риск, держатель регистрационного удостоверения должен оценить необходимость внесения изменений в информацию о лекарственном препарате и предложить контекст соответствующих изменений.
Заключение должно включать предварительные предложения по оптимизации или дальнейшей оценки соотношения польза-риск с целью их последующего обсуждения с соответствующими регуляторными органами. Данные предложения могут включать меры минимизации риска.
Для лекарственных препаратов, имеющих план по фармаконадзору и минимизации риска предложения должны быть включены в план по фармаконадзору и план минимизации риска.
2.2.20. Раздел ПОБ «Приложения к ПОБ»
ПОБ должен включать следующие приложения:
- Справочная информация
- Кумулятивные обобщающие табличные данные по серьезным нежелательным явлениям, выявленным в ходе клинических исследований/испытаний
- Кумулятивные и интервальные обобщающие табличные данные по серьезным и несерьезным нежелательным реакциям по данным пострегистрационного применения
- Табличные данные по сигналам
- Оценка сигналов, если применимо
- Перечень всех пострегистрационных исследований по безопасности
2.3 Система качества ПОБ на уровне держателя регистрационных удостоверений
Держатель регистрационного удостоверения должен иметь сформированные структуры и процессы для подготовки, контроля качества, обзора и представления ПОБ, включая контроль испонения в процессе и после их оценки. Данные структуры и процессы должны быть описаны в письменных процедурах системы качества держателя регистрационного удостоверения.
Процессы фармаконадзора включают ряд направлений, которые могут оказать непосредственное влияние на качество ПОБ (например, обработка сообщений о нежелательных реакциях, полученных в рамках спонтанного репортирования или клинических исследований; обзор литературы; выявление, валидация и оценка сигнала; дополнительные меры по фармаконадзору и пострегистрационной исследовательской деятельности; процедуры обработки и объединения данных при оценке пользы и риска и иные). Система качества должна описывать взаимосвязь между процессами, каналы информирования и обязанности по процедурам сбора всей имеющей отношение информации для включения в ПОБ. Должны быть разработаны и внедрены документированные процедуры по контролю качества процессов с целью обеспечения полноты и точности данных, представляемых в ПОБ. Важность интегрированной оценки соотношения польза-риск определяет необходимость обеспечения вклада различных департаментов/отделов при подготовке ПОБ.
ПОБ должен содержать оценку специальных запросов по аспектам профиля безопасности со стороны регуляторных органов. У держателя регистрационного удостоверения должен иметься механизм, обеспечивающий надлежащую обработку и ответы на запросы регуляторных органов.
Представление обобщающих табличных данных должно подвергаться процедуре верификации данных в отношении баз данных держателя регистрационного удостоверения с целью обеспечения точности и полноты представляемых данных в отношении нежелательных реакций/явлений. Процессы размещения запросов в базе данных, используемые параметры для извлечения данных и контроль качества должны быть надлежащим образом документированы.
Надлежащая система качества держателя регистрационного удостоверения должна исключить риск невыполнения держателем регистрационного удостоверения требований законодательства, таких как:
а) непредставление отчета: полное непредставление ПОБ, нарушение графика или сроков подачи ПОБ (без предварительного согласования с регуляторными органами);
б) необоснованное непредставление запрошенной информации;
в) низкое качество отчетов (плохое документирование либо недостаточная информация или оценка представлена по новой информации по безопасности, сигналы по безопасности, оценка риска, оценка пользы и интегрированный анализ соотношения польза-риск, отсутствие указание о неправильном применении, отсутствие стандартной терминологии, необоснованное исключение случаев, непредставление информации по факторам риска);
г) представление ПОБ без отражения ранее полученных запросов от регуляторных органов.
Все значимые отклонения от процедуры подготовки и представления ПОБ должны быть документированы и соответствующие корректировочные и предупредительные мероприятия приняты. Данная документация должна быть доступна в любое время.
В случае делегирования обязанностей по подготовке ПОБ третьим сторонам, держатель регистрационного удостоверения должен обеспечить наличие у третьей стороны надлежащей системы качества, соответствующей требованиям законодательства.
2.4. Обучение персонала процедурам по ПОБ
Ответственностью лица уполномоченного по фармаконадзору, является обеспечение надлежащей квалификации, опыта и обучения персонала по фармаконадзору, оценке медицинской информации и контроля качества, задействованных в процедурах подготовки, обзора, контроля качества, оценки и представления ПОБ. При необходимости выполняется необходимое обучение по различным составляющим процессам, аспектам знаний и навыкам. Направления обучения должны включать аспекты законодательства, руководств, научную оценку данных, письменные процедуры по аспектам подготовки ПОБ. Документирование процесса обучения должно подтверждать его прохождение до начала выполнения соответствующих функций по ПОБ.
2.5. Порядок представления ПОБ
2.5.1. Стандартный порядок представления ПОБ
Периодичность и сроки представления периодических отчетов по безопасности лекарственных препаратов определяются согласно перечню, утверждаемому регуляторными органами государств-членов ЕАЭС.
Для лекарственных препаратов, международное непатентованное наименование или группировочное наименование которых не включены в указанный перечень, периодичность представления ПОБ составляет:
-каждые 6 месяцев от международной даты регистрации на протяжении первых 2 лет;
— ежегодно на протяжении последующих 2 лет;
— далее — каждые 3 года.
Срок подачи ПОБ от даты окончания сбора данных составляет не более 90 календарных дней.
2.5.2. Внеочередная подача ПОБ
ПОБ подлежит подаче незамедлительно, в срок до 60 календарных дней, от даты получения письменного запроса регуляторного органа государства-члена ЕАЭС.
2.5.3. Форма подачи ПОБ
ПОБ подлежат подаче в электронном виде с возможностью текстового поиска на русском языке или английском языке с обязательным переводом на русский язык следующих разделов: краткого изложения основного содержания, интегрированного анализа соотношения польза-риск по одобренным показаниям и заключения. По запросу регуляторного органа держатель регистрационного удостоверения обязан в течении 30 календарных дней представить перевод на русский язык других разделов ПОБ.